
A comprehensive map of mobile element insertions from 5,675 genomes
Recently, the research groups of Prof. XU Tao and Prof. HE Shunmin from the Institute of Biophysics of the Chinese Academy of Sciences reported the genome resource NyuWa of mobile element insertions (MEIs), in order to promote MEI genetic and medical research in world population. This study was published in Nucleic Acids Research (Figure 1).

Figure 1. The paper published in Nucleic Acids Research
In the human genome, Alu, LINE-1 (L1), SINE-VNTR-Alu (SVA), and HERV-K are the families of mobile elements that are generally considered to be still active and capable of forming new insertions in the genome through transposition, known as Mobile Element Insertion (MEI). Transposition events have the potential to interrupt functional regions of the genome, disrupting normal gene function, affecting transcript expression or splicing, and leading to disease. More than 120 human genetic diseases have been reported to be associated with transposon-mediated insertions, including hemophilia, Dante's disease, neurofibromatosis, and cancers. In addition to effects via insertion events, the intrinsic sequence properties of the transposable elements confer functional effects on the host for some MEIs, making MEIs qualitatively different from other typical structural variants. At the same time, the preference of MEI integration sites has long been a focus of researchers. The distribution of these sites is not uniform and is influenced by various factors such as DNA sequence and chromatin environment.
Despite the important functions of MEIs, there is a paucity of resources for integrating polymorphic transposable elements in the human genome, which is the basis for phenotype-variant association analysis. in 2017, the Thousand Genomes Project conducted a comprehensive analysis of MEIs in 2504 genomes, identifying over 20,000 polymorphic MEI loci. Watkins et al. extended the findings based on the 1,000 Genomes dataset by analyzing the variant profile of MEIs in a global population using 296 genomic data from the Simons Genome Diversity Project. However, the genetic resources for these MEIs are mainly from European populations. Even in gnomAD-SV, the largest cohort of structural variation studies to date, only 1304 samples are from East Asia. Since the Chinese Han Chinese are the most populous ethnic group in East Asia and worldwide, MEI studies and resources for Chinese populations are scarce.
This study systematically analyzed the genomic distribution, mutation characteristics, and functional impact of MEIs at the population level, and constructed a comprehensive MEI repository, especially the MEI map for the Chinese population. This work is part of the NyuWa Chinese Population Genome Project led by Academician XU Tao and Researcher HE Shunmin from the Institute of Biophysics, Chinese Academy of Sciences. Previously, the NyuWa Genome Project has already published the Chinese population genetic variation atlas and reference panel, as well as the Chinese Population Genome Repository (http://bigdata.ibp.ac.cn/NyuWa/) to lay the foundation for genetic and medical research in the Chinese population.
The authors systematically identified MEIs by combining 2998 high-depth whole-genome sequencing data from the Nuwa genome resource and 2677 low-depth whole-genome sequencing data from the Thousand Genomes Project. On average, more than 1000 MEI variants were detected per individual, the majority of which were insertions of Alu components (Figure 2).
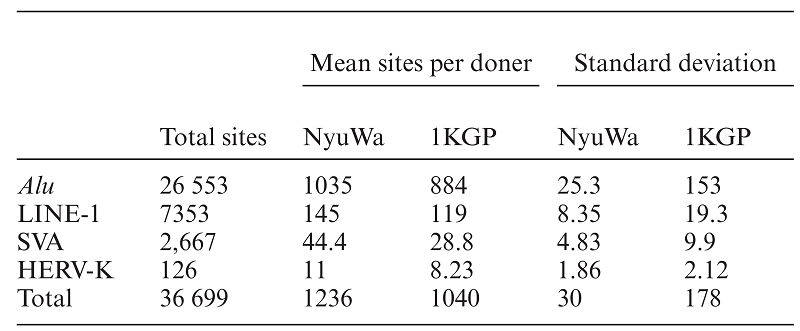
Figure 2. The MEIs detected in this study.
The authors analyzed the chromosome distribution of MEIs and found that L1 insertion was significantly enriched in the region near the centromeres (Figure 3). The enrichment of L1 insertion variants in the vicinity of the centromere DNA may be due to the high number of α-satellite sequences in the vicinity of the mitotic DNA, and the relatively low GC content is more favorable for L1 insertion. On the other hand, considering that the active transposons in the neo-transposon region identified in previous studies may contribute to the nascent of the centromere, the authors suggest that the enrichment of L1 in the centromere region may also be biologically important. This finding needs to be investigated in subsequent studies.
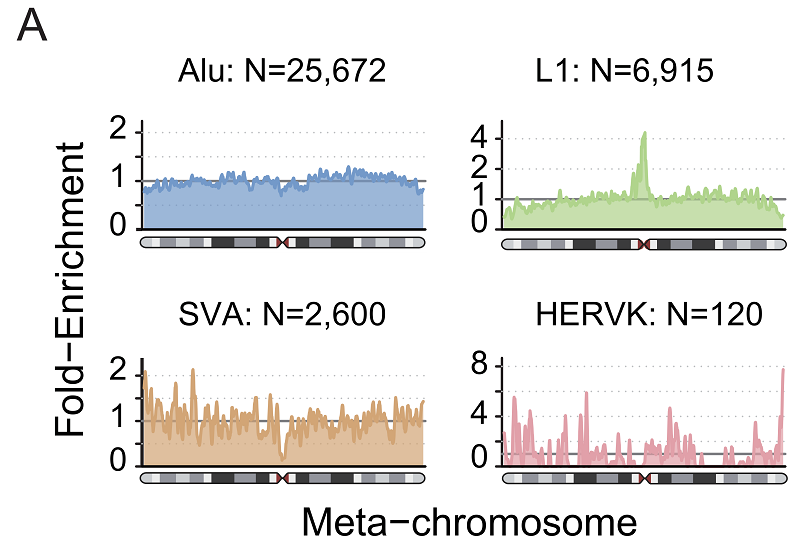
Figure 3. MEI density in "meta-chromosome".
Next, the authors estimated the mutation rate (per bp per generation) of MEI in the two data sets ("Nuwa" and 1,000 genomes) separately, 1.609x10-11 for "Nuwa" and 1.464x10-11 for 1,000 genomes - very close results. The results are very similar, with approximately 1 new MEI event per 16-17 births. Furthermore, by comparing MEI diversity and SNP heterozygosity in different populations, the authors found a high correlation, with African populations having the highest MEI diversity and SNP heterozygosity (Figure 4).
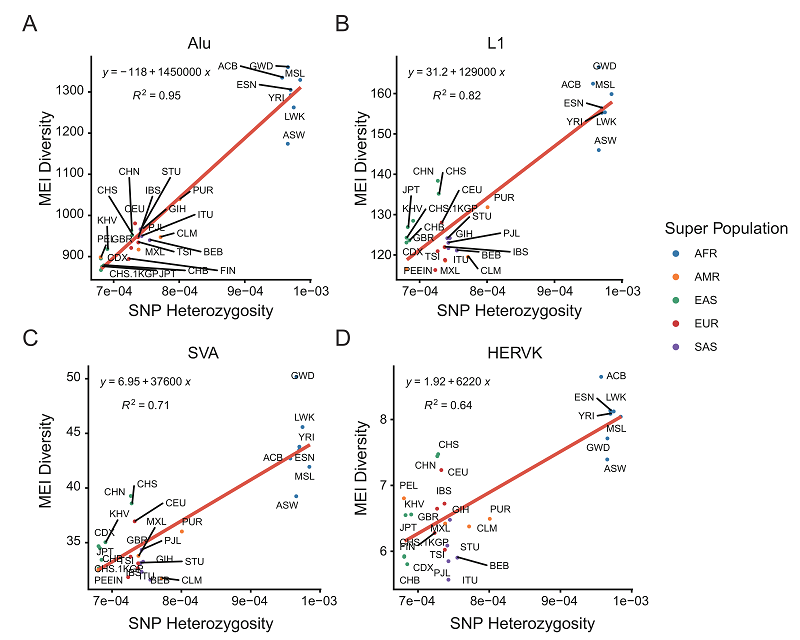
Figure 4. Correlation between SNP heterozygosity and MEI diversity.
Theoretically, MEIs in the protein-coding region can cause loss of gene function by interrupting the open reading frame. After functional annotation of MEIs, the authors found that everyone contains an average of 24 MEIs that truncate proteins (Figure 5). Combining short variants (SNP and InDel) with other structural variants, MEIs contribute approximately 9.4% of the truncated protein variants per individual. This result demonstrates the importance of including MEIs in routine analysis of genome-wide data.
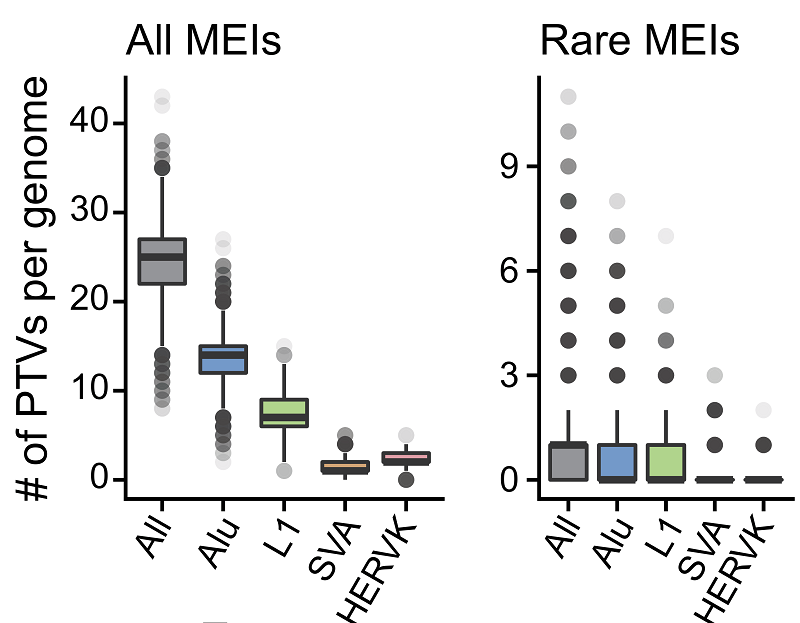
Figure 5. Box plots of counts of predicted PTVs by MEI
The insertion of L1 is usually accompanied by 3' transduction, i.e. the sequence downstream of its original 3' end is inserted into the new site along with the L1. Based on this feature, the authors analyzed the source-offspring relationship of L1, identified some new source-offspring pairs, found some potentially active L1 loci, and discovered differences in their distribution in different populations (Figure 6).
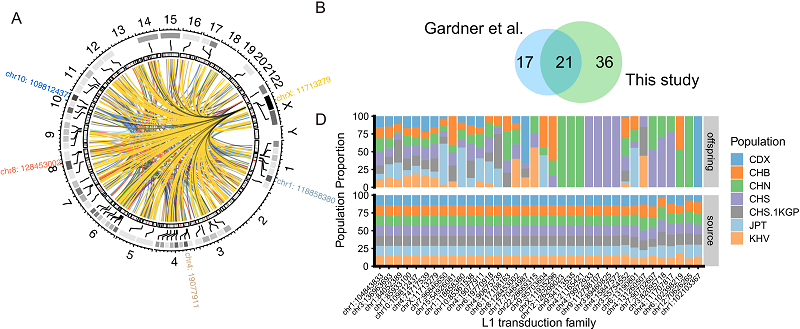
Figure 6. L1 3' transduction
Finally, to facilitate the search and use by researchers of removable components, the authors have constructed an open database, HMEID, to include the MEIs identified in this study at: http://bigdata.ibp.ac.cn/HMEID/. In addition, this database is also part of the "Nuwa" genomic data resource (http://bigdata.ibp.ac.cn/NyuWa_variants/).
In summary, the authors reported a comprehensive map of 36,699 non-reference MEIs constructed from 5,675 genomes, including 2,998 Chinese samples (~26.2X, NyuWa) and 2,677 samples from the 1000 Genomes Project (~7.4X, 1KGP). The insertion of L1 was found to be highly enriched in the region of the centromere regions, implying a possible role of the chromosomal environment in transposable element insertion. After functional annotation, the authors estimated that MEI contributes to 9.3% of the protein truncation events in each individual. Finally, the authors have created a companion database called HMEID for public use. This resource represents the most recent and largest genome-wide study on MEI to date, and it is expected that it will play a role in exploring new knowledge of human MEI.
Link to the paper:https://doi.org/10.1093/nar/gkac128
Contact: HE Shunmin
Institute of Biophysics, Chinese Academy of Sciences
Beijing 100101, China
Email: heshunmin@ibp.ac.cn
(Reported by Dr. HE Shunmin's group)