
New technology enables high-throughput, high-resolution structural analysis of proteins in cells
Cryo-electron microscopy single particle techniques have been achieved to resolve the structures of proteins in solution to near atomic resolution, and even to atomic resolution on apoferritin. However, proteins in their working state cellular environment may have more native conformations and may form more complete complexes than purified proteins, so achieving high-resolution in situ protein structure is an important goal in cryo-EM.
Cryo-electron tomography (cryo-ET) combined with subtomo-averaging is currently the main tool to solve in situ protein structures, but the reconstruction of tomogram requires the collection of tilt series, which severely reduces the data collection throughput, and the alignment errors and sample distortion making it difficult to reach high resolution.
In order to achieve high-throughput and high-resolution structure determination of proteins in situ, ZHANG Xinzheng (Institute of Biophysics, Chinese Academy of Sciences) has been working on developing new methods that is not based on cryo-ET to solve protein structures in situ. Our paper entitled "Determining protein structures in cellular lamella at pseudo-atomic resolution by GisSPA" which is based on the isSPA methods published in 2021 and accelerated by GPU, was published online by Nature Communications on March 15, 2023. GPU acceleration improves computational efficiency by about 400-500 times than the earlier CPU codes.
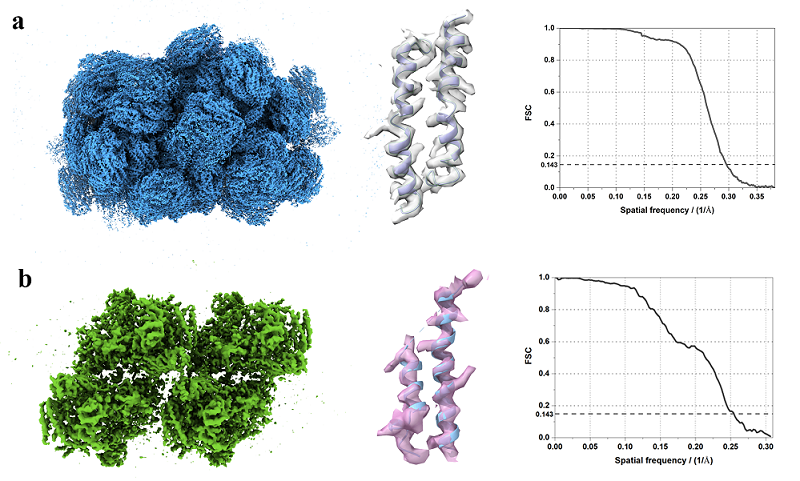
Figure 1. The high-resolution in situ structures and their map densities presentation: structure of PBS (a) and structure of PSII (b)
In this work, we applied GisSPA to resolve the high-resolution structures of phycobilisome with a molecular weight of about 14.7 MD and photosystem II complexes with a molecular weight of about 1.5 MD in cellular lamellae with a resolution of 3.4 Å and 3.9 Å, respectively (Figure 1). In addition, the range of applicability of the GisSPA algorithm for proteins of different molecular weight sizes and slice thicknesses was explored in this work, and it was concluded that the smallest resolvable complex molecular weight is about 1.1 MD at sample thicknesses of 100-150 nm (Figure 2). GisSPA improves the in-situ data collection efficiency by 30-40 times compared to that of cryo-ET, and the resolution of target protein is also significantly improved.
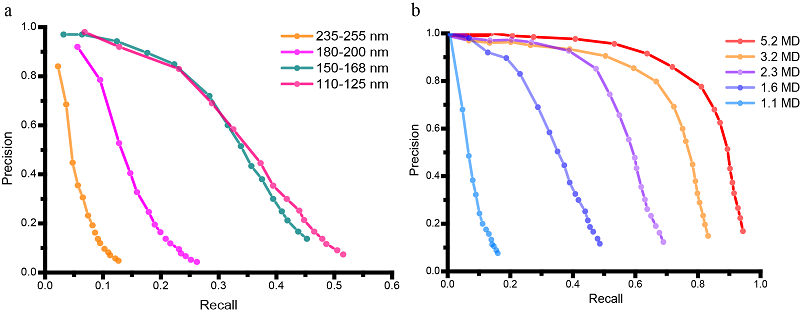
Figure 2. Precision-recall curves describing the detection efficiency of GisSPA on samples with different thickness and molecular weights.
ZHANG Xinzheng (Institute of Biophysics, Chinese Academy of Sciences) and WAN Xiaohua (Beijing University of Technology) are the co-corresponding authors of this paper. CHENG Jing, from ZHANG Xinzheng's lab, and LIU Tong, a graduate student at the Institute of Computing, Chinese Academy of Sciences, are the co-first authors of this paper. The project was funded by the National Key R&D Program of China, the National Natural Science Foundation of China, the Strategic Priority Research Program of the Chinese Academy of Sciences and the Key Research Program of Frontier Sciences at the Chinese Academy of Sciences.
The FIB lamellae used in the paper are from SUI Senfang's lab at Tsinghua University. Sui's team has applied cryo-ET and the isSPA methods to study the PBS-PSII-PSI-LHC giant complex of red algae, in which the isSPA methods improved the resolution of the complex to 3.3 Å. The work was published online on the same day in Nature, entitled "In situ structure of the red algal phycobilisome-PSII-PSI-LHC megacomplex", with SUI Senfang and WANG Hongwei from Tsinghua University and ZHANG Xinzheng from the Institute of Biophysics, Chinese Academy of Sciences as co-corresponding authors. YOU Xin from SUI Senfang's lab, ZHANG Xing from WANG Hongwei's lab, CHENG Jing from ZHANG Xinzheng's lab and XIAO Yanan from SUI Senfang's lab are co-first authors.
Professor ZHANG Xinzheng has been working on the development of novel in situ structure resolution techniques, based on which a protein recognition algorithm was developed earlier and the optimal weighting function was derived, and the new weighting function significantly improved the detection efficiency of target proteins in overlapping densities. A sorting method to reduce the false positives to effectively mitigate the model bias was introduced in earlier work, thus enabling the high-resolution reconstruction, the work was published in The Innovation in 2021 (https://doi.org/10.1016/j.xinn.2021.100166). A more extensive deduction of the methods is related to a paper published in Biophysics Reports (https://doi.org/10.52601/bpr.2021.210001).
Paper links:
Determining protein structures in cellular lamella at pseudo-atomic resolution by GisSPA
https://doi.org/10.1038/s41467-023-36175-y
In situ structure of the red algal phycobilisome-PSII-PSI-LHC megacomplex
https://doi.org/10.1038/s41586-023-05831-0
Contact: ZHANG Xinzheng
Institute of Biophysics, Chinese Academy of Sciences
Beijing 100101, China
Email: xzzhang@ibp.ac.cn
(Reported by Dr. ZHANG Xinzheng's group)