
The Third Achievement of NyuWa Genome Resource: Genome-wide Microsatellite Variation Map for Chinese Population
On April 12, 2023, the Institute of Biophysics, Chinese Academy of Sciences made a significant breakthrough in short tandem repeat (STR) research. Professor Xu Tao's team and professor He Shunmin's team co-authored an article on their latest finding, which was published in the esteemed international journal, Nature Communications (Figure 1). This research is a vital component of the NyuWa Genome Project, spearheaded by Xu Tao and He Shunmin.

Figure 1 The article published in Nature Communications
The NyuWa Genome Project has set out to establish a comprehensive genome-wide data resource and conduct systematic analyses of genetic variation for the Chinese population. To achieve this, the research teams have accomplished two significant milestones: Firstly, they have published a variation map containing SNP/Indel and loss-of-function variants in coding and noncoding genes, as well as the first deep whole-genome sequencing-based reference panel for the Chinese population (Cell Reports, 2021, "NyuWa Genome resource: A deep whole-genome sequencing-based variation profile and reference panel for the Chinese population"). Secondly, the research teams have systematically analyzed whole genome sequencing data from 5,675 individuals (including 2,998 Chinese individuals from the NyuWa Genome resource) and constructed a mobile element insertion map for various populations (Nucleic Acids Research, 2022, "Characterizing mobile element insertions in 5,675 genomes").
Recently, the third achievement of the NyuWa Genome Project is focused on short tandem repeats (STRs; also known as microsatellites). STRs are one to six base pairs repeats and account for only approximately 3% of human genomes. However, approximately 60 STRs have been associated with human Mendelian diseases, including ataxia, amyotrophic lateral sclerosis, Huntington disease, frontotemporal dementia, and various neurological disorders.
STRs are primarily characterized by the repetitive structure, which endows STRs a higher mutation rate than other parts of the genome. Most mutations of STRs are due to expansions or contractions of repeat units, resulting in digital length polymorphisms. Emerging evidence has shown that many polymorphic STRs (pSTRs) can regulate various molecular and cellular processes such as DNA methylation, gene expression, and alternative splicing, ultimately affecting complex human traits.
The lack of large-scale studies on STR variation in human and the challenges of STR variation analysis have hindered researches on STR-related human traits and diseases. Therefore, one urgent need is to construct a full and accurate catalog of pSTRs in the human genome.
The latest achievement of the NyuWa Genome Project focuses on addressing these challenges. This work has constructed a genome-wide STR variation map for populations worldwide, including the Chinese population, conducted a systematic analysis about the genome distribution, mutation patterns, functional properties, gene-regulatory effects, population characteristics, and population differences of STRs, and built a comprehensive STR variation resource.
The research teams analysed the high-coverage whole genome sequencing data of 3,983 genomes from the NyuWa Genome resource and 2,504 genomes from the 1000 Genomes Project to identify genome-wide STR variation. After rigorous quality screening, over 1.55 million alleles at 366,013 polymorphic STR (pSTR) loci were identified. Of these, approximately 1/3 (523,063 alleles) were specifically identified in the NyuWa dataset (Figure 2).
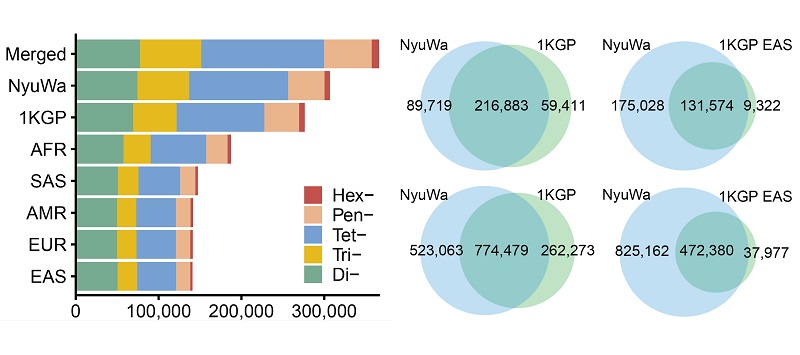
Figure 2 Number of pSTR loci and pSTR alleles identified in this work
Using the pSTR call set, the research teams analyzed the mutational patterns of STR loci and found that STR mutations were influenced by motif length, chromosome context and epigenetic features. They also observed that hexameric pSTRs were enriched within subtelomeric regions, while no such bias was found for other types of pSTRs or mSTRs (Figure 3).
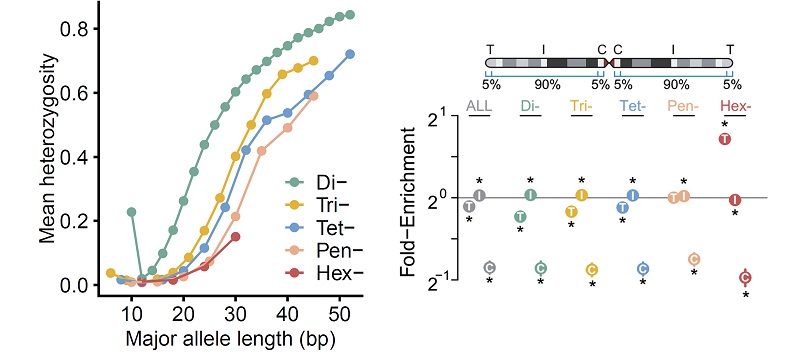
Figure 3 Mutation pattern of pSTRs
To explore the potential gene-regulatory effect of pSTRs, the research teams identified 3273 and 1117 pSTRs whose repeat numbers were associated with gene expression and 3'UTR alternative polyadenylation, respectively. They also found that these pSTRs were more concentrated in regions with active histone marks and open chromatin (Figure 4).
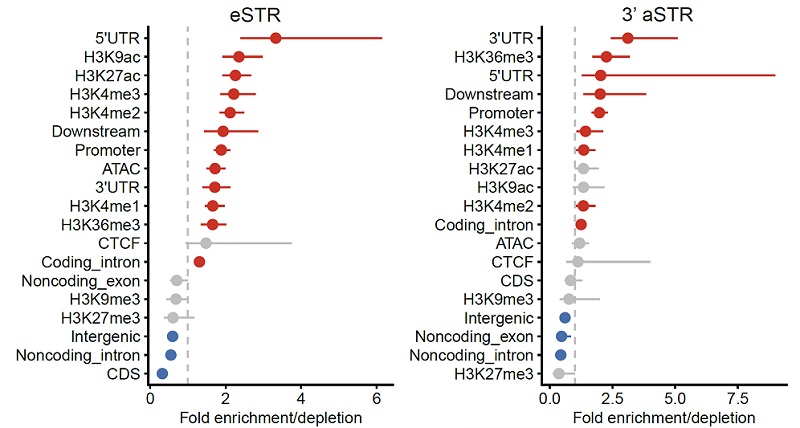
Figure 4 Enrichment of eSTRs and 3'aSTRs in genomic regions
The research teams have identified several pSTRs with significant mean length differences between superpopulations, which may contribute to phenotypic differentiation between different populations. For instance, a pSTR in the intron of UBE2L3, a member of the E2 ubiquitin-conjugating enzyme family, was mostly expanded in individuals from East Asia. This pSTR was in strong LD with several GWAS SNPs implicated in multiple phenotypes, such as Crohn's disease and systemic lupus erythematosus (Figure 5).
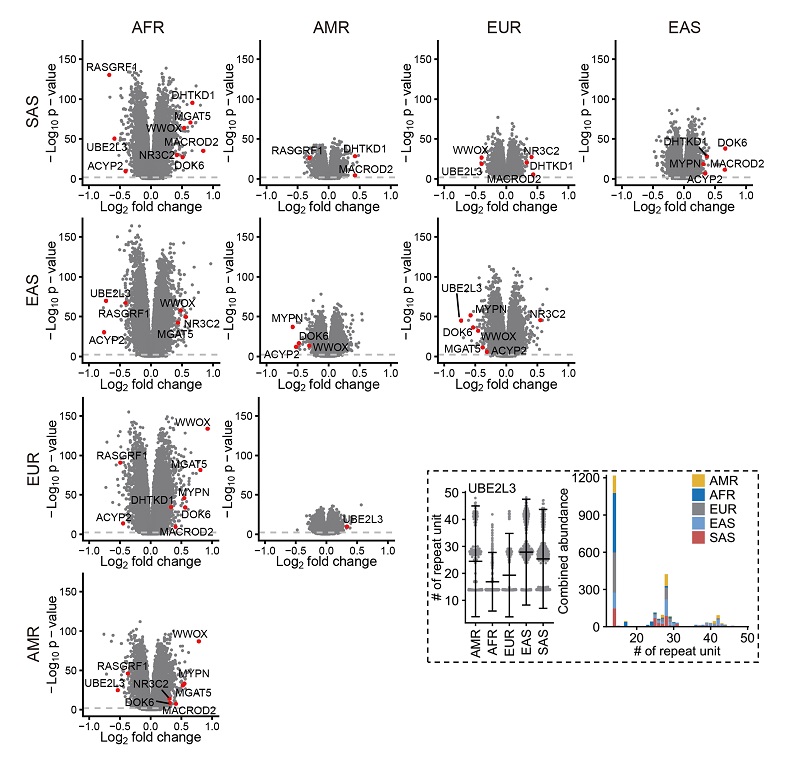
Figure 5 pSTRs with significant mean length differences between superpopulations
In summary, the study presents a comprehensive variation map of 366,013 pSTR loci for 6,487 genomes, comprising 3,983 Chinese samples (~31.5x, NyuWa) and 2,504 samples from the 1,000 Genomes Project (~33.3x, 1KGP). The research teams investigated the factors that influence STR mutations, including motif length, chromosome context and epigenetic features. They identified some pSTRs with potential gene-regulatory effects, including 3273 eSTRs and 1117 3'aSTRs, and found that these STRs were enriched in regulatory elements and accessible chromatin regions. They also studied the population characteristics of STRs and identified differential STRs between and within populations. In addition, they provided the allele distributions of 60 known disease-causing STRs.
This study represents one of the largest and latest genome-wide studies of STR variation in various populations, which will promote our understanding of the diversity and potential function of STRs in the human genome.
Professor He Shunmin and Professor Xu Tao from the Institute of Biophysics, Chinese Academy of Sciences are the co-corresponding authors of this paper. Shi Yirong and Niu Yiwei from the Institute of Biophysics, Chinese Academy of Sciences are the co-first authors of this paper. The research was supported by Strategic Priority Research Program of the Chinese Academy of Sciences, the National Natural Science Foundation of China, the National Key R&D Program of China, the 14th Five-year Informatization Plan of the Chinese Academy of Sciences, and the National Genomics Data Center. This research is expected to provide a basis and reference for future studies on STR variation in human genomes.
Original article link: https://doi.org/10.1038/s41467-023-37690-8
Contact: HE Shunmin
Institute of Biophysics, Chinese Academy of Sciences
Beijing 100101, China
Email: heshunmin@ibp.ac.cn
(Reported by Dr. HE Shunmin's group)