
High-throughput analysis of the three-dimensional structure of protein machines in complex biological environments
On September 8, 2021, the journal The Innovation published online the research result of ZHANG Xinzheng's group at the Institute of Biophysics, Chinese Academy of Sciences, "Determining structures in a native environment using single-particle cryo-electron microscopy images". This work follows the block-based reconstruction algorithm developed in 2018 (Nat Commun 9, 1552) for structure refinement in protein stacked densities, and extends the algorithm to in situ structure resolution. The algorithm proposes a new method for protein molecules in complex biological environments, utilizing a tilt-free data acquisition approach, optimizing the protein detection algorithm and removing model bias to obtain the target protein structure, resulting in a significant increase in data collection throughput by more than tenfold, as well as an increase in resolution.
Cryo-electron tomographic (cryo-ET) reconstruction is currently used for structural analysis in complex biological environments. Cryo-ET requires tilting the sample stage to collect tilting series data at different angles, thus the throughput is very low, and more than 10 days of data acquisition is often required to achieve a high resolution reconstruction, which is unaffordable for most platforms. Second, the resolution of in situ structure resolution based on tomographic reconstruction is very limited due to the lack of data and the endogenous defects of tomographic reconstruction techniques. These shortcomings have severely limited the development of in situ structure analysis by cryo-electron microscopy.
For proteins in complex biological environments, the density of the target protein overlaps with the density of other surrounding proteins. A protein detection algorithm was developed based on this, and the optimal weights of the detection function were derived. The new function we derived improved the efficiency of detecting the target protein in the overlapping density. Combined with the sorting function to reduce the false positive data introduced by the protein detection algorithm, the model bias problem is effectively mitigated and high-resolution reconstruction is achieved (Figure 1). The algorithm has achieved high-throughput high-resolution structures of the following samples: membrane proteins bound to liposomes, glycoproteins in non-icosahedral viruses, and protein complexes in subcellular units.
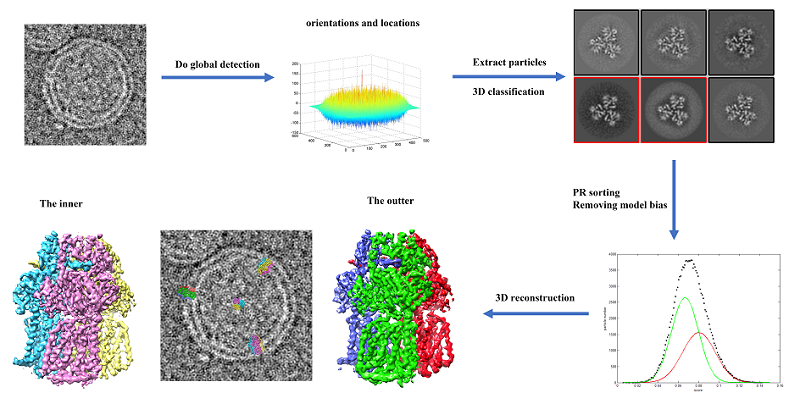
Figure1. Reconstruction of membrane proteins bound to liposomes
ZHANG Xinzheng, a researcher at the Institute of Biophysics, Chinese Academy of Sciences, is the corresponding author of this paper, CHENG Jing, a doctoral student in ZHANG Xinzheng's group, is the first author of this paper, and LI Bufan, a doctoral student in ZHANG Xinzheng's group, and SI Long, a doctoral student in CHANG Wenrui/LI Mei's group at the Institute of Biophysics, Chinese Academy of Sciences, also contributed to this research. This work was supported by the project of National Natural Science Foundation of China and other grants.
Paper link: https://www.sciencedirect.com/science/article/pii/S2666675821000916
Contact: ZHANG Xinzheng
Institute of Biophysics, Chinese Academy of Sciences
Beijing 100101, China
Email: xzzhang@ibp.ac.cn
(Reported by Dr. ZHANG Xinzheng's group)