
Researchers reveal molecular mechanism of host innate immunity modulating γ-herpesvirus replication
γ-herpesviruses, including Kaposi's Sarcoma-associated herpesvirus (KSHV) and Epstein-Barr virus (EBV), are the etiological agents for Kaposi's sarcoma, primary effusion lymphoma, nasopharyngeal carcinoma and other malignant tumors. The life cycle of a γ-herpesvirus has two phases: a quiescent latent phase and a replicative lytic phase. The balance between these two phases is critical for γ-herpesvirus to establish life-long infection in host and cause diseases. Therefore, comprehensive understanding of the mechanisms regulating the switch between latency and lytic replication of γ-herpesvirus is important to designing effective strategies to control virus infections.
To date, most studies focused on the role of viral proteins, such as RTA and LANA, and host epigenetic regulators in regulating the switch of KSHV life cycle, while not much is known about the role of host innate immunity in this process. Previous studies did demonstrate that interferons, as important antiviral cytokines of innate immunity, could inhibit lytic replication of KSHV, and deficiency in interferons led to higher lytic reactivation from latency, indicating that interferons play important roles in modulating lytic replication and latent infection. However, the mechanisms underlying the action of interferons remained unclear.
Recently, a research group led by Prof. Deng Hongyu from the Institute of Biophysics of the Chinese Academy of Sciences provided an answer to this question by uncovering a novel mechanism adopted by RNF213, an interferon-stimulated gene (ISG), to inhibit KSHV infection and lytic reactivation. As a downstream effector protein of the interferon pathway, RNF213 serves as an E3 ubiquitin ligase and promotes ubiquitination and degradation of the KSHV "molecular switch" protein RTA. This study was published in The Proceedings of the National Academy of Sciences, USA (PNAS).
By screening an ISGs library, the researchers found that RNF213 inhibited murine γ-herpesvirus 68 (MHV-68) replication. Further studies demonstrated that RNF213 suppressed viral early gene transcription and viral genome replication by down-regulating the expression of RTA, which is a "molecular switch" for MHV-68 life cycle. Importantly, de novo infection and lytic reactivation of KSHV were also inhibited by RNF213 through decreasing the expression of the homologous KSHV "molecular switch" protein RTA.
Mechanistically, the researchers showed that RNF213 degraded both MHV-68 and KSHV RTA proteins through the proteasome but not the lysosome pathway. RNF213 interacted with RTA directly and functioned as an E3 ubiquitin ligase to ubiquitinate RTA via K48 linkage.
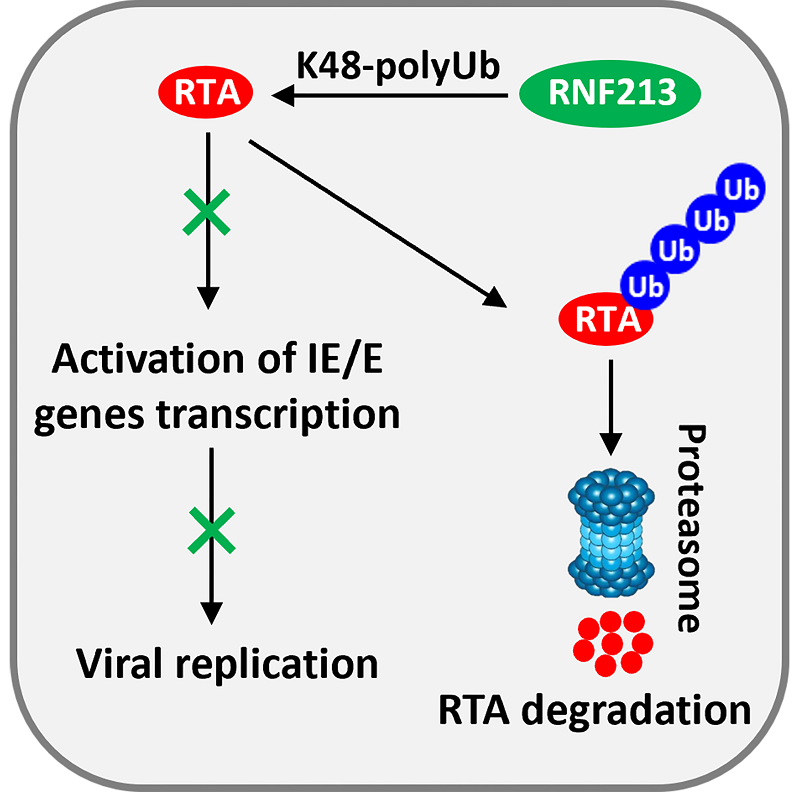
Figure. Model for the mechanism of RNF213 inhibiting γ-herpesvirus replication
(Image by Dr. DENG Hongyu's group)
In summary, this study uncovered that the ISG product RNF213 serves as an E3 ubiquitin ligase and inhibits the de novo infection and lytic reactivation of γ-herpesviruses by degrading RTA through the ubiquitin-proteasome pathway. The findings helps us to better understand how the host innate immunity, especially the interferon pathway, modulate γ-herpesvirus infection, and provides new insights into measures to combat viral infections.
Article link: https://www.pnas.org/doi/10.1073/pnas.2218825120
Contact: DENG Hongyu
Institute of Biophysics, Chinese Academy of Sciences
Beijing 100101, China
Email: hydeng@ibp.ac.cn
(Reported by Dr. DENG Hongyu's group)