
"NyuWa" Genome Project Releases Fifth Achievement
Recently, the "NyuWa" genome team has elaborated on the impact of non-coding regulatory elements on phenotypic evolution under adaptive selection, with related research paper published in Molecular Biology and Evolution. This work is part of the "NyuWa" Chinese population genome project led by Prof. XU Tao and Prof. HE Shunmin from the Institute of Biophysics, Chinese Academy of Sciences.
The "NyuWa" Chinese population genome project aims to construct a comprehensive genome data resource for the Chinese population and thoroughly analyze genetic variations in the Chinese population genome to support research on diseases and precision medicine among the Chinese population. Prior to this, based on large-scale whole-genome sequencing data of the Han Chinese population, the NyuWa genome project mapped the genome-wide variation spectrum of SNPs, InDels, STRs, and MEIs in the Chinese Han population and also analyzed recent adaptive selection in the Han Chinese population.
The recently released work further focuses on the evolution of the population genome in less-studied non-coding regulatory regions. In this study, the research team systematically identified genome-wide adaptive selection and revealed the impact of adaptive selection of non-coding regulatory elements on the population's phenotype.
The research findings indicate that approximately 12% of the autosomal genome regions of the Han Chinese population are influenced by adaptive selection, with around 15% of non-coding regulatory elements (promoters and enhancers) undergoing adaptive selection. This proportion is slightly lower than that of protein-coding genes.
Through functional enrichment analysis of target genes of regulatory elements experiencing adaptive selection, it was found that positively selected regulatory elements are mainly enriched in biological pathways such as cell adhesion, while balancing selection regulatory elements are mainly enriched in immune-related pathways.
This study further located archaic gene introgression regions subject to positive selection, and found that such events are concentrated in the 3p13.11 region, with regulatory elements in this region mainly related to adaptation to ultraviolet radiation. The key target gene in this region is the HYAL gene, encoding hyaluronidase to degrade hyaluronic acid.
Furthermore, the research team analyzed the impact of positively selected sites on linked disease risk genes and found that almost half of the disease risk genes are rapidly eliminated under positive selection, while the efficiency of clearing the other half of the disease risk genes under positive selection is reduced.
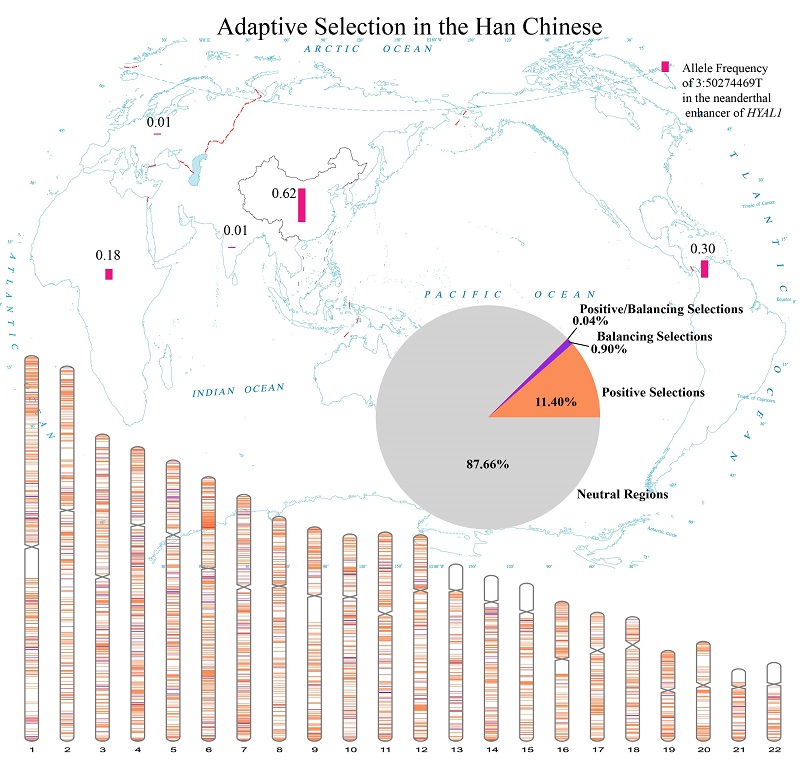
Fig. Reported by Center for Big Data Research in Health
Article link:https://doi.org/10.1093/molbev/msae034
Contact: HE Shunmin
Institute of Biophysics, Chinese Academy of Sciences
Beijing 100101, China
Email: heshunmin@ibp.ac.cn
(Reported by Prof. HE Shunmin's group)